MRA - mutation resistance analyzer
This service is new! Although we simulated several test data, we cannot guarantee for perfect performance at the moment.
In case of any problems or suggestions do not hesitate to contact us (hans.kestler [at] uni-ulm.de).
Supplementary information
Algorithm
The alignment algorithm used in the analyzer is published here: Extended pairwise local alignment of wild card DNA/RNA sequences using dynamic programming
Test sequences
( Link to the MRA )
Just copy one of these sequences to the corresponding form field of the analyzer.
Detailed Usage

The sequence data must be submitted to the server by using the corresponding form field. The sequence can be given required in FASTA-Format or as plain text. An optional identifier can be used to be shown in the result view. You can either copy the sequence in the text area or upload it in FASTA-format with a valid file extension (fasta).
Select the gene / protein-sequence, you want the sequence to align to.
Always check the checkbox for confirming that you have read and accepted the "Terms of use".
If you use the "worst case calculation" modus, wild bases will aligned to the worst possible mutation. Please note, this calculation will take more time!
The submitted data is compared via SWAT to the reference sequence. Only sequences sharing more than 60 nucleotids with the reference sequence will be analysed. Access to the information is granted by a special id, so you can share your results with other users. Results can also be explicitly deleted in the result view.
Important: Everyone who may guess the id will have access to your results, so make sure not providing sensitive data e.g. name of patients.
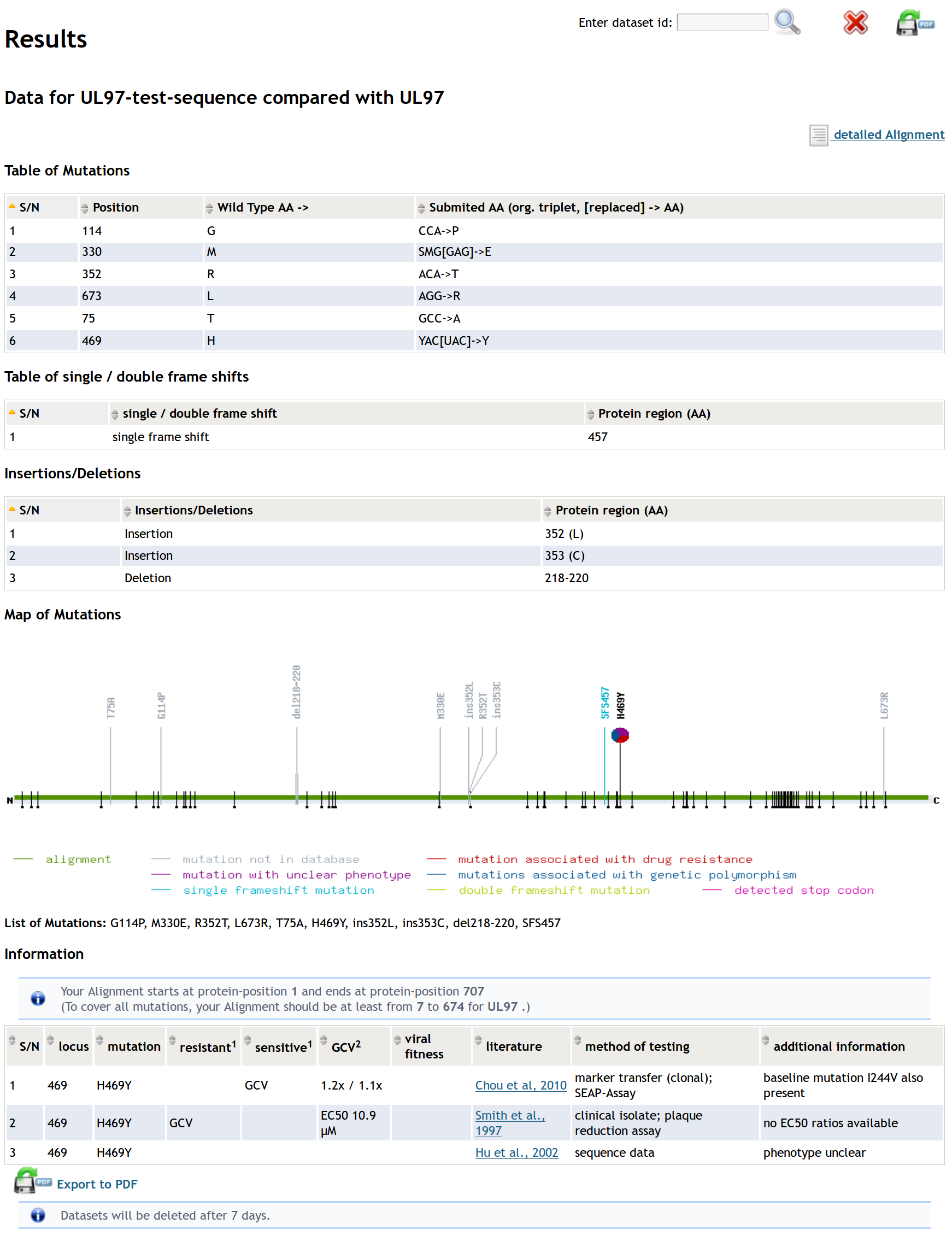
In the result view mutations, insertions, deletions, frameshifts and stop-codons are listed in tables in the upper area. Positions of mutations are visualized in the plotting area. If additional information about a mutation is found in our databases, it is indicated by a pie chart representing the (different) hypothesis of the publication(s) and it is also shown in the lower table.
Hint: Enabled Javascript to have access to sorting and filtering forms of the table.
You can permanently delete the results by clicking the "Delete" button on the top right of the result view. Otherwise, the results will be deleted after 7 days.
You can export the results in PDF-format by clicking the "Export" button on the top right of the result view.
Hans A. Kestler
University of Ulm
D-89069 Ulm, Germany